
BRET experiments reveal the complex spectrum of 5-HT2AR signaling in living cells
To gain insight into the molecular determinants that drive ligand-mediated 5-HT2AR signaling bias, we studied the engagement of the receptor’s proximal effectors (G proteins and β-arrestins) upon stimulation with structurally closely related probes of the endogenous agonist 5-HT (Fig. 1), including Met-I (3-(2-aminoethyl)−1-methyl-1H-indol-5-ol hydrochloride), Nitro-I (2-(5-nitro-1H-indol-3-yl)ethamine hydrochloride), OTV1 (2-[5-(2,3-dihydro-1,4-benzodioxin-6-yl)−1H-indol-3-yl]ethan-1-amine) and OTV2 (2-(5-phenoxy-1H-indol-3-yl)ethan-1-amine)). The two latter compounds have been obtained from a virtual screen (see method section). Competition binding experiments with [3H]-ketanserin confirm that the test compounds bind to the orthosteric binding site of the 5-HT2AR (Fig. 1).
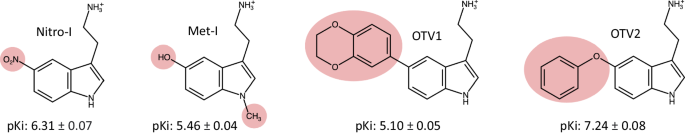
Ligand binding affinities (pKi) are indicated for Nitro-I, Met-I, OTV1 and OTV2 obtained in [3H]ketanserin competition binding experiments in CHO cells (n = 3). The data represent the mean ± SD (see methods section).
In a first screen, we explored the coupling spectrum of the 5-HT2AR to several Gα protein subtypes and β-arrestins upon stimulation with Nitro-I, Met-I, OTV1 and OTV2 in living HEK-293 cells using BRET-based assays (Fig. 2A). The pEC50, Emax and log τ/KA for Gαq, Gα11, Gα14, Gα15 Gαi1, Gαi2, Gαi3, GαoA, GαoB, and Gαz are listed in Table 1, whereas full concentration response curves are shown only for representative members of the Gαq and Gαi family and β-arrestin 1 and 2 in Fig. 2B–G. We find that all compounds are full agonists toward the canonical Gαq pathway, but only partial agonists on the Gαi family and βarrs when compared to 5-HT. Notably, among the Gαi family members, the relative efficacy is greater toward Gαi1 vs Gαi2, or Gαi3. To further characterize the signaling profiles promoted by the 5-HT analogs, the transduction coefficient (log τ/KA) was determined for each pathway using the operational model13,14 followed by calculation of the ligand-physiology bias7 factor between pathways using the endogenous agonist 5-HT as the reference.
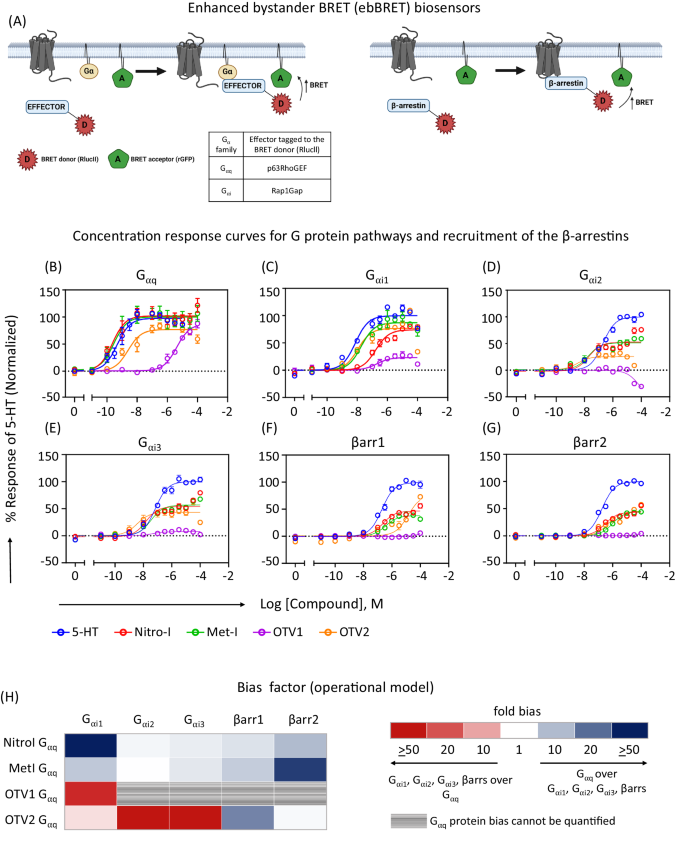
A Schematic representation of enhanced bystander BRET (ebBRET) biosensors used to measure G protein activation and β-arrestin (βarr) recruitment in HEK-293 cells, created with BioRender. B–G Dose response curves depicting the activity of Nitro-I, Met-I, OTV1 and OTV2 at the different G protein signaling pathways and for the recruitment of βarr 1 and 2. The ligand-promoted BRET (ΔBRET) was further normalized with respect to the response of 5-HT (mean ± SEM; n = 3). H Bias factor for the Gαq, Gαi family and βarr1 and 2. The operational model was used for bias calculations (see method section in supplementary information).
Figure 2H illustrates the coupling preference and physiology-bias profile promoted by each of the compounds as compared to 5-HT. Nitro-I and Met-I show a general physiology bias for the canonical Gαq over Gαi1, Gαi2, Gαi3, β-arrestin 1, and 2 (light to dark blue, Fig. 2H). Of note is the high magnitude of Gαq bias over β-arrestin 2 for both compounds with 10 (Nitro-I) and > 50 fold (Met-I), and the high Gαq bias over Gαi1 with > 50 (Nitro-I) and 8-fold bias (Met-I).
Interestingly, introducing an extended substituent (i.e. phenoxy substituent) in position 5 of the indole fragment (Fig. 1), as in OTV2, results in a distinct coupling (Fig. 2B–G) and bias profile of 5-HT2AR (Fig. 2H) compared to Nitro-I and Met-I. OTV2 induces a general bias toward the Gαi family (Gαi1, Gαi2, Gαi3) over Gαq (light to dark red colors, Fig. 2H) in comparison to 5-HT, which is mainly driven by the increased potency of OTV2 towards the Gαi family members. Furthermore, we observe that OTV2 has a substantially reduced ability to promote the recruitment of β-arrestin 1, yielding a 17-fold Gαq bias over β-arrestin 1 while the Gαq bias over β-arrestin 2 observed for Nitro-1 and Met-I was lost for OTV2 (Fig. 2H and Table 1).
Further extension of position 5 (i.e. 1,4-benzodioxin) (Fig. 1) produces the most dramatic changes in the coupling (Fig. 2B–G) and physiology-bias profile (Fig. 2H). OTV1 loses to a large extent its stimulating activity for Gαi2, Gαi3 as well as its ability to recruit β-arrestin 1 and 2 (Fig. 2D–G). This results in a very significant Gαq activation preference over Gαi1, with a bias factor > 20 compared to 5-HT. An even greater preference is observed toward Gαq vs Gαi2, Gαi3, β-arrestin 1 and 2. However, virtually no activation of these pathways prevented us from calculating a formal bias factor (gray colored, Fig. 2H). Whether this preference results from a structurally-driven signaling bias or from the lower potency of OTV1 toward all pathways (i.e. activation of pathways that are not strongly coupled to the receptor are difficult to be detected) cannot be determined.
An interesting observation is that some of the observed pEC50 values are many orders of magnitude larger than the corresponding binding affinities (Fig. 1B). An example is Met-I with a pEC50 of 9.6 for Gαq activation (Table 1) versus a pKi of 5.5 for its binding affinity to the 5-HT2AR (Fig. 1). This difference most likely results from the diverse experimental setups including the expression level of receptors or G proteins15. Interestingly, additional BRET experiments show that receptor expression levels do not affect the observed pEC50 values (Fig. S9). In contrast, we find that increasing levels of Gαq significantly increment the apparent potency of Met-I (Fig. S10). This finding demonstrates that the expression level of G protein is a critical parameter that largely contributes to the differences observed between pEC50 of G protein activation and pKi of ligand binding. It further underscores the difficulties associated with comparing data points across distinct experimental setups and could also explain the differences observed between cell-based and ex vivo experiments.
Met-I and OTV1 reduce the basal activity of the Gαi1 pathway via the 5-HT2AR in postmortem brain tissue
Next, we explored the ability of our molecular probes to modulate the activity of diverse G protein subtypes (Gαq/11, Gαi1, Gαi2, Gαi3) in postmortem human brain tissue. For this, we used [35S]GTPγS binding experiments coupled to immunoprecipitation of Gα subunits of heterotrimeric G proteins (Fig. 3A) in postmortem human dorsolateral prefrontal cortex (PFC) membrane-enriched fractions16,17. To confirm that these effects were mediated through 5-HT2AR, the same assays were carried out in the presence of MDL-11,939, a 5-HT2AR-selective neutral antagonist18 (Fig. 3B–E, Fig. S1, Table 2).
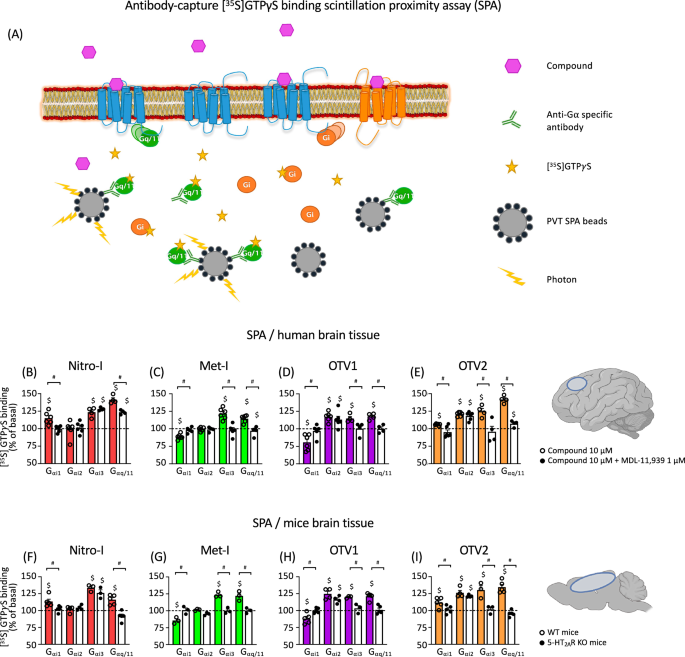
A Schematic representation of the SPA methodology, created using BioRender. SPA allows the determination of the activation level of different Gα subunit subtypes present in postmortem brain tissue thanks to the selective immunoprecipitation of each of them and their coupling to protein A-coated polyvinyltoluene SPA beads. Modulation of specific [35S]GTPγS binding to Gαi1, Gαi2, Gαi3 and Gαq/11 proteins by 10 μM Nitro-I (B), Met-I (C), Otava 3575001 (OTV1) (D) and Otava 3736689 (OTV2) (E) in human prefrontal cortex both in the absence (colored bars) and in the presence (white bars) of the 5-HT2AR antagonist MDL-11,939. Basal values of specific [35S]GTPγS binding to the different G proteins are expressed as 100% and stimulatory/inhibitory effects on the respective basal are shown. Individual dots represent different assays 4–6 carried out for each subunit/condition performed in duplicate or triplicate. White dots represent data for the drug-alone condition, and black dots represent data for the drug + MDL condition (B–E). #p < 0.05 vs 100%; $p < 0.05 vs incubation in the presence of 1 µM MDL-11,939 (t-test). Modulation of specific [35S]GTPγS binding to Gαi1-, Gαi2-, Gαi3– and Gαq/11 proteins by 10 μM Nitro-I (F), Met-I (G), OTV1 (H) and OTV2 (I) in brain cortex tissue from WT (colored bars) and 5-HT2AR KO mice (white bars). #p < 0.05 vs 100%; $p < 0.05 WT vs 5-HT2AR KO (t-test). Basal values of specific [35S]GTPγS binding to the different Gα proteins are expressed as 100%, and stimulatory/inhibitory effects are expressed as % of the respective basal activity. Individual dots represent the different assays (3–5) for each subunit performed in duplicate or triplicate. White dots represent data for WT mice and black dots represent data for KO mice (F–I).
Our study reveals that Nitro-I triggered statistically significant activation of Gαi1, Gαi3 and Gαq/11. Among them, only Gαi1 and Gαq/11 are 5-HT2AR-mediated, as co-incubation with MDL-11,939 reversed the observed effect completely or partially, respectively (Fig. 3B, Table 2). Met-I modulated the activity of all studied Gα subunit subtypes, with the exception of Gαi2. Selective 5-HT2AR inhibition with MDL-11,939 suggests that inverse agonism at Gαi1 and agonism at Gαi3, Gαq/11 are directly mediated by 5-HT2AR (Fig. 3C, Table 2). In the same way, although OTV1 and OTV2 are able to modulate all studied Gα subunit subtypes (Fig. 3D, E, Table 2), only Gαi1, Gαi3 and Gαq/11 modulation is 5-HT2AR-mediated, but not Gαi2 modulation. However, one main difference is related to the observation that Met-I and OTV1 elicit inverse agonism at the Gαi1 whereas Nitro-I and OTV2 show a Gαi1 agonism. The overall observed Gαq/11 activation for tested compounds in postmortem brain samples (Fig. 3B–E) is in line with the activation of the canonical Gαq in our BRET experiments in living cells (Fig. 2D). Interestingly, differences are found for the regulation of Gαi1, Gαi2 and Gαi3 subunit’s activity between postmortem brain samples and the cell-based setup. These differences are not surprising considering variations in the experimental environment between postmortem brain samples and cell-based assays. This includes different expression levels of G proteins, 5-HT2AR and other GPCRs. In fact, the presence of other GPCR types has been reported to promote the formation of heteromers which can alter the coupling response of 5-HT2AR. For instance, signaling via the CB1R-5-HT2AR heteromer promotes Gαi coupling and not the canonical Gαq coupling of the 5-HT2AR19. The presence of other factors in the postmortem tissue vs the cell line system could also explain the difference. The fact that Met-I and OTV1 are inverse agonists on Gαi1 in the human brain but partial agonists for this pathway in the cell-based assays could be related to a higher basal tone for the 5-HT2AR-promoted Gαi1 activation in the tissue.
To further demonstrate the role of 5-HT2AR in the observed effects in postmortem human brains, tissue homogenates from the brain cortex of wild type (WT) and 5-HT2AR knockout (KO) mice were incubated with Nitro-I, Met-I, OTV1, and OTV2 in [35S]GTPγS binding experiments (Fig. 3F–I, Fig. S2, Table S1). Importantly, these experiments confirm 5-HT2AR-mediated signaling profiles observed in postmortem human PFC. Only small differences are found for the Nitro-I-induced Gαq/11 activation, which is exclusively mediated through the 5-HT2AR in mice, whereas a partial blockade was observed in human brain with the 5-HT2AR-selective antagonist MDL-11,939.
All in all, our experiments highlight the complex signaling profile elicited by the tested compounds (Fig. 3B–I) that behave as agonists, inverse agonists or show no effect for the different subunit subtypes in homogenates from human PFC or mice brain cortex. Furthermore, we find that the observed coupling profile is often the result of interaction with multiple receptor types, as selective 5-HT2AR antagonism does not always reverse the observed effects. This is also indicated by the finding that some of the effects are still present in 5-HT2AR KO mice brain tissue. Most importantly, we identify two compounds, Met-I and OTV1, that show an inverse agonism effect over the Gαi1 via the 5-HT2AR. Since different guanosine diphosphate (GDP) concentrations and specific antibodies are used for the detection of each Gα subunit, results obtained from this methodological approach are semiquantitative. Thus, no quantitative comparisons can be made between the different studied subunits, no bias factor can be calculated, and only the effects of different compounds over the same subunit can be compared.
Gαi agonism is implicated in psychosis-related effects
To interrogate the implication of specific 5-HT2AR-mediated pathways in psychosis-related behavior, we investigated the effects of in vivo administration of our test compounds in mice on the head twitch response (HTR). The HTR serves as a behavioral proxy in rodents for human psychedelic effects, and can be used to discriminate hallucinogenic and non-hallucinogenic 5-HT2AR agonists9,20,21,22,23,24,25. Thus, increasing doses of Nitro-I, Met-I, OTV1 or OTV2 were administered intracerebroventricularly (ICV), as well as DOI, a classic psychedelic 5-HT2AR agonist, chosen as control. In a first step, we confirmed that our test compounds are not lethal or produced any physiological or neurotoxicity symptoms in mice for tested doses by the Irwin test (see Methods).
As expected, the HTR is significantly increased by our reference compound DOI with respect to vehicle-treated animals (Fig. S3, inset). We found that Nitro-I and OTV2, compounds triggering a 5-HT2AR-mediated Gαi1, Gαi3 and Gαq/11 agonism in postmortem brain assays, significantly increased HTR as compared to vehicle administration (Fig. S3A, D). Importantly, this effect is absent in 5-HT2AR KO mice (Fig. 4A, D), indicating a 5-HT2AR-dependent mechanism, and supporting other data showing that the 5-HT2AR is necessary for the expression of HTR21.
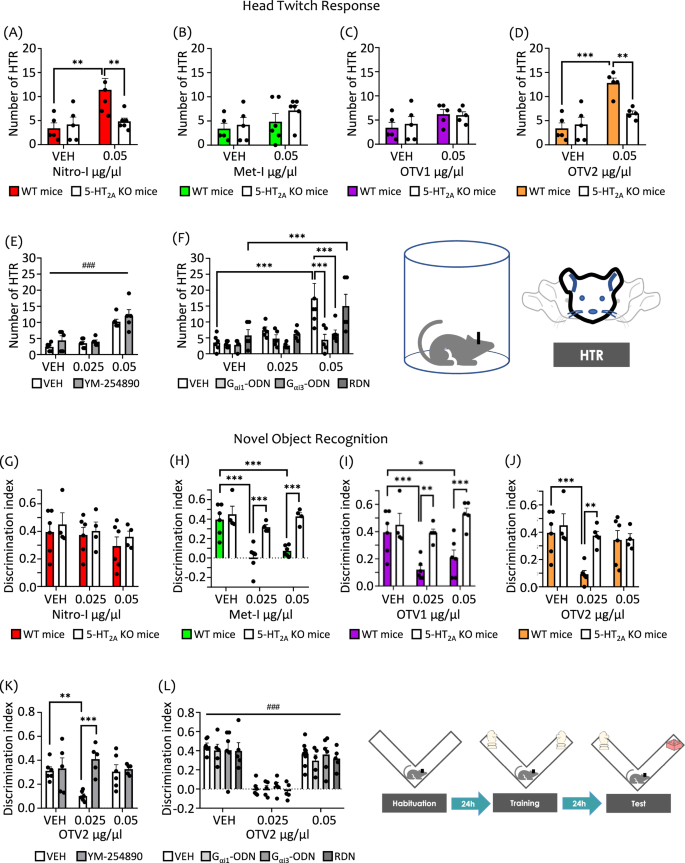
HTR in wild-type (WT) and 5-HT2AR knockout (KO) mice following ICV administration of (A) Nitro-I, (B) Met-I, (C) OTV1, and (D) OTV2, or vehicle (VEH). The increase in HTR induced by Nitro-I and OTV2 at the dose of 0.05 µg/µl was absent in KO mice. E ICV administration of YM-254890 (16 µM) did not modulate the increase in HTR induced by OTV2 at the dose of 0.05 µg/µl. F ICV administration of specific antisense oligonucleotides (ODN), Gαi1-ODN, Gαi3-ODN, but not control random oligonucleotides (RDN) blocked HTR induced by OTV2 at the dose of 0.05 µg/µl. NOR memory test in WT and 5-HT2AR KO mice following ICV administration of (G) Nitro-I, (H) Met-I, (I) OTV1, and (J) OTV2 or vehicle (VEH). In WT mice, Met-I and OTV1 induced memory deficits at the dose of 0.025 and 0.05 µg/µl, and OTV2 was effective only at the dose of 0.025 µg/µl. These effects were abrogated in KO mice. Nitro-I did not trigger memory deficits at any of the doses tested. K YM-254890 (16 µM) abrogated the memory deficits induced by OTV2 at the dose of 0.025 µg/µl. L ICV administration of Gαi1-ODN, Gαi3-ODN, or the control RDN sequence did not modulate the memory deficits induced by OTV2 at the dose of 0.025 µg/µl. The data represent mean ± SEM. The number of mice used in the experiments (n) corresponds to the individual points in the graph. **p < 0.01, ***p < 0.001, and ###p < 0.001 (main effect of treatment). Two-way ANOVAs followed by Fisher’s post-hoc test.
Interestingly, Met-I and OTV1, which showed 5-HT2AR-mediated Gαi1 inverse agonism in brain tissue experiments, do not increase HTR at any of the doses tested (Fig. S3B, C) in WT or 5-HT2AR KO mice (Fig. 4B, C). Note that in addition to inverse agonism of Gαi1, both compounds also stimulate the activity of Gαi3 and Gαq subtypes. To evaluate the possible involvement of Gαi1, Gαi3 and Gαq in the regulation of psychosis-related effects (i.e. HTR) through 5-HT2AR stimulation, we used two different methodological approaches. On one hand, we carried out pharmacological inhibition of Gαq/11 by ICV administration of YM-254890. On the other hand, we reduced the expression level of Gαi1 and Gαi3 genes, GNAI1 and GNAI3 respectively, by the ICV administration of specific antisense oligonucleotides (ODNs). OTV2 was selected as a model drug for these experiments, as it induces HTR and shows a 5-HT2AR-mediated activation of Gαi1, Gαi3 and Gαq in postmortem brain tissue.
Importantly, we find that OTV2-induced HTR at the dose of 0.05 µg/µl was not modulated by Gαq/11 inhibition using YM-254890 (Fig. 4E). Instead, decreasing protein levels for both Gαi1 and Gαi3 using ODNs abrogated the OTV2-induced HTR (Fig. 4F). We confirmed that Gαi1 and Gαi3 protein levels were significantly decreased in mice after chronic treatment with Gαi1– or Gαi3-ODNs compared to ODN-RDN (i.e., a random oligo) using Western blot analysis (Fig. S4), whereas no change was observed for the expression levels of Gαq/11. The specificity of the used antibodies has been previously demonstrated16. Surprisingly, although the used ODNs had been previously described in the literature26, a cross-effect of Gαi1 ODN and Gαi3 ODN treatment over both Gαi1 and Gαi3 protein expression levels was observed in our hands (Figure S4). Therefore, we are not able to discriminate between Gαi1– and Gαi3– mediated effects with the currently existing reagents.
Altogether, our results provide evidence that the activation of Gαi protein family-coupled signaling pathways (Gαi1 and/or Gαi3) via the 5-HT2AR is a main contributor to psychosis-related effects in mice. Although our data suggest that Gαi1-activation is necessary for this effect, we cannot completely exclude mechanisms other than Gαi/o activation in mediating HTR. Previous studies describe the involvement of other coupling partners including Gαq25,27,28,29,30, although there are also studies showing that Gαq KO mice inhibit only partially HTR27, suggesting additional contributing mechanisms in HTR. Moreover, studies have reported the involvement of Gαs proteins30, Gβγ subunits31, and β-arrestins11,12 in HTR.
Long-term memory performance is linked to 5-HT2AR-induced Gαq activation
To investigate the ability of our compounds to modulate cognitive performance via the 5-HT2AR, we carried out the novel object recognition (NOR) test in WT and 5-HT2AR KO mice. Indeed, we find that Met-I and OTV1 induce significant long-term memory deficits in WT mice at both doses tested through a 5-HT2AR-dependent mechanism (Fig. 4H, I). Interestingly, OTV2 produces long-term memory deficits in WT mice only at the lower dose, and this effect is absent in KO mice (Fig. 4J). To our surprise, we find that Nitro-I is the only compound that does not induce long-term memory deficits (Fig. 4G).
To interrogate which pathway is associated with these effects over cognition, we chose again OTV2, our model compound able to induce long-term cognitive impairment in addition to HTR. For discriminating which of the Gα protein subtypes (Gαi1, Gαi3 and Gαq) activated by OTV2 through a 5-HT2AR-mediated mechanism in human and mouse brain tissue are implicated in these memory effects, we used again the two approaches previously described in the HTR section: (i) reduction of protein expression levels of Gαi1 and Gαi3 via ODN administration and (ii) Gαq pharmacological inhibition using YM-254890.
We find that reducing the protein levels of Gαi1 and Gαi3 via ODNs did not influence long-term memory deficits induced by OTV2 at the dose of 0.025 μg/μl (Fig. 4L). Instead, inhibiting Gαq activation with YM-254890 abrogated OTV2-induced long-term memory deficits (Fig. 4K). These results provide first evidence that long-term memory performance is linked to modulation of the Gαq-coupled pathway via the 5-HT2AR. Importantly, our data suggest further that cognitive deficits require other co-factors/events in addition to Gαq activation. This can be concluded from the observation that although Nitro-I also activates Gαq in cell-based and brain tissue experiments, it does not elicit cognitive deficits.
In a final experiment, we also evaluated whether our compounds induced anhedonia, one of the features of depression, and our results showed that none of the doses administered acutely evoked this behavior in WT or KO mice (Fig. S5).
Ligands with differential 5-HT2AR coupling profiles and in vivo responses establish distinct ELC2 interactions
In the previous experiments, we have employed signaling probes that are structurally closely related to the endogenous neurotransmitter 5-HT. One main difference between these signaling probes are diverse substituents in position 5 of the indole scaffold, which proved to alter the 5-HT2AR coupling profile in living cells (Fig. 2), and in postmortem brain tissue (Fig. 3) and behavioral responses (Fig. 4).
Molecular dynamics (MD) simulations have been shown to be a valuable tool for interrogating structural and dynamic events linked to GPCR function32,33,34 including signaling bias35. Here, we exploited this approach to elucidate the structural determinants that may be responsible for the different in vitro and in vivo responses of the 5-HT2AR. For this, we constructed 3D structural models of the complexes by docking all ligands into the orthosteric binding site of the 5-HT2AR and subjected each complex to MD simulation. Structural inspection of the most representative clusters found in simulations reveals that the positioning of the main tryptamine scaffold of all compounds resembles the experimentally solved tryptamine pose of serotonin in the 5-HT1AR (PDB id 7E2Y) with the following interactions of high contact frequencies (Fig. 5A, B): (i) a salt bridge with D3.32 and (ii) a hydrophobic sandwich formed of V3.33, F6.51 and F6.52. In fact, these positions have been corroborated by numerous mutational studies for tryptamine, 5-hydroxytryptamine and other closely related derivatives (i.e., V3.3336, F6.51, and F6.5237,38,39). With this central scaffold in place, extension in position 5, as observed in OTV1 (1,4 benzodioxin) and OTV2 (phenoxy), are oriented towards the extracellular loop 2 (ECL2) where they result in increased contact frequencies (Fig. 5C). This interaction is mediated by hydrophobic interactions, in particular with L228 and L229, but also by additional hydrogen bonds with the backbone of L229 in the case of OTV1. The role of ECL2, specifically L229, mediating interactions with bulkier compounds in serotonin receptors has been reported by Wacker et al.40.
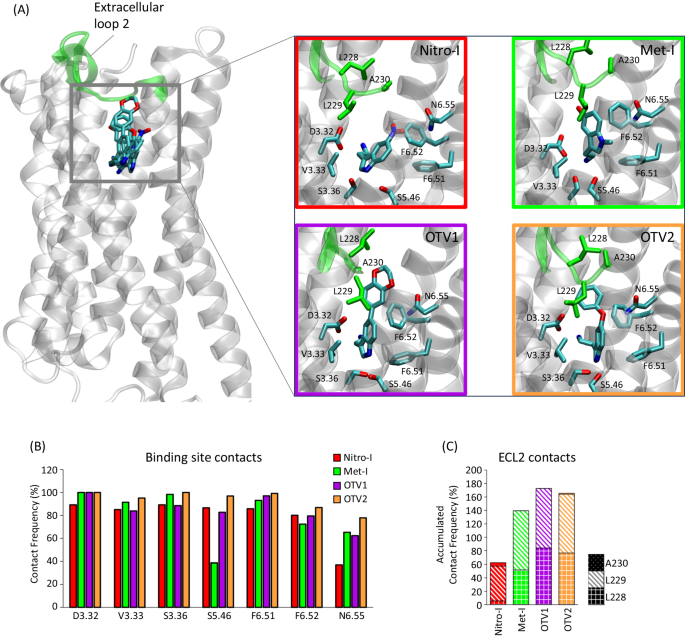
A 3D model of the ligand-5-HT2AR interactions for Nitro-I, Met-I, OTV1, and OTV2. The ECL2 and included residues are highlighted in green. B The contact frequency (%) of residues in the transmembrane region of the receptor for each ligand. Contact frequencies have been computed for the main conformational cluster (see Methods) extracted from three replicates of 500 ns MD simulations (3 × 500 ns). C Accumulated contact frequency of residues in the ECL2 for each ligand computed for the main cluster extracted from three replicates of 500 ns MD simulations (3 × 500 ns).
Interestingly, we observe that differential ligand-receptor interactions are associated with distinct ligand binding affinities at the 5-HT2AR in cell-based assays (Fig. 5D). The highest affinity for 5-HT2AR is found for OTV2 which exposes a phenoxy substituent in position 5 of the indole fragment. Elongation of this position to a benzodioxan dramatically reduces ligand binding affinity as seen for OTV1. In addition, we observe that introducing a methyl substituent at the heteroaromatic nitrogen in position 1 is not favorable for ligand binding affinity, as observed for Met-I. One could speculate that ligand binding affinities are correlated with the total amount of receptor contacts (i.e. pocket plus ECL2 contacts). However, this is not the case as specific contact types (e.g. hydrogen bonds or Van der Waals) contribute differently to the binding affinity which is not taken into account when computing the total number of contacts.
Nevertheless, the type of substitution in position 5 of the indole scaffold and corresponding ECL2 contacts seem to alter 5-HT2AR coupling in living cells (Fig. 2). Compounds with small-sized substitutions in position 5 (i.e., Nitro-I and Met-I) show overall Gαq physiology-bias (compared to 5-HT) over the tested Gαi proteins as well as β-arrestin 1 and 2 (Fig. 2H). Changes are observed upon extension of position 5. Both, OTV1 (5-benzodioxin substituent) and OTV2 (5-phenoxy substituent) show a Gαi1 bias over the canonical Gαq protein (Fig. 2H). Our structural models of ligand binding suggest that this change in coupling profile could be related to increased interaction with the ECL2 via the extended substituent in position 5 (Fig. 5C).
Structural modifications of the indole scaffold also significantly impact the 5-HT2AR-induced G protein activation profile in human brain tissue: Nitro-I (Gαi1, Gαq/11 agonism), Met-I (inverse Gαi1 agonism and Gαi3, Gαq/11 agonism), OTV1 (Gαi1, Gαi3, Gαq/11 agonism) and OTV2 (inverse Gαi1 agonism and Gαi3, Gαq/11 agonism). For instance, our data suggest that increased ECL2 contacts (Met-I, OTV1, OTV2, Fig. 5C) promote Gαi3 activation in brain tissue (Fig. 3C–E) compared to Nitro-I (Fig. 3B). One of the most important structural observations is that the regulation of the psychosis-associated Gαi1 pathway can be mediated by diverse mechanisms. On one hand, we find that differential interaction within the ECL2 (Fig. 5A, C) can convert a Gαi1 agonism of OTV2 (hydrophobic ECL2 interaction via 5-phenoxy) (Fig. 3E) into a Gαi1 inverse agonism as observed for OTV1 (polar ECL2 interaction via 5-benzodioxan) (Fig. 3D). In addition, we find that Gαi1 agonism can also be induced by compounds with a relatively low number of ECL2 contacts (Nitro-I, Figs. 3B, 5C).
Based on this finding and our structural models, we propose that Gαi1 agonism does not require strong ECL2 contacts, whereas Gαi1 inverse agonism can be induced via differential interactions with the ECL2. In fact, this is in line with structural observations for the hallucinogenic compound LSD (PDB 6WGT) that stimulates Gαi/o coupling9 compared to the non-hallucinogenic compound lisuride (PDB 7WC7) with no Gαi/o coupling through 5-HT2AR9. Both compounds are structurally closely related with a highly similar binding mode but show differences in their interaction frequencies with the ECL2 in MD simulations (Fig. S6). Finally, another relevant structural observation is that compounds that induce cognitive deficits (Met-I, OTV1 and OTV2) are characterized by pronounced contacts with the ECL2 when compared to Nitro-I.
Overall, our structural insights can have important implications for the rational design of drug candidates with a tailored signaling profile and in vivo response applied to the treatment of psychiatric diseases.


{kind=link}